README.md
In hw538/cfDNAPro: cfDNAPro extracts and Visualises biological features from whole genome sequencing data of cell-free DNA
cfDNAPro 




Official tutorials
For detailed documentation please visit: https://cfdnapro.readthedocs.io/en/latest/
Declaration
cfDNAPro is designed for research only.
Challenges in the cfDNA fragment length calculation
Unlike genomic DNA, cfDNA has specific fragmentation patterns. Ambiguous definition of "fragment length" by various alignment software is raising concerns: see page 9 footnote in SAM file format spec: https://samtools.github.io/hts-specs/SAMv1.pdf
Cell-free DNA data fragmentomic analysis requires single-molecule level resolution, which further emphasizes the importance of accurate/un-biased feature extraction.
cfDNAPro is designed to resolve this issue and standardize the cfDNA fragmentomic analysis using the bioconductor R ecosystem.
Input
cfDNAPro is specifically written for cell-free DNA paire-ed whole-genome sequencing data.
Its ensures accurate (i.e. up-to-standard) calculation of fragmentomic features (e.g., fragment lengths and motif)
Output
cfDNApro can extract (i.e., "quantify in a standandised and robust way") these features/bio-markers:
- fragment length
- fragment start/end/upstream/downstream motifs
- copy number variation
- single nucleotide mutation
- more...
Feature extration depends on essential data objects/R packages in the Bioconductor ecosystem, such as Rsamtools, plyranges, GenomicAlignments, GenomeInfoDb and Biostrings.
Data engineering depends on packges in the tidyverse ecosystem, such as dplyr, and stringr.
All plots depend on ggplot2 R packge.
For issues/feature request etc., please contact:
Author: Haichao Wang
wanghaichao2014@gmail.com
Author: Paulius D. Mennea
paulius.mennea@cruk.cam.ac.uk
Nitzan Rosenfeld Lab admin mailbox:
Rosenfeld.LabAdmin@cruk.cam.ac.uk
Quick Start 1
Read in bam file, return the fragment length counts.
A straightforward and frequent user case: calculate the fragment size of a bam file, use the following code:
# install cfDNAPro newest version
if (!require(devtools)) install.packages("devtools")
devtools::install_github("hw538/cfDNAPro", build_vignettes = FALSE)
# calculate insert size of a bam file
library(cfDNAPro)
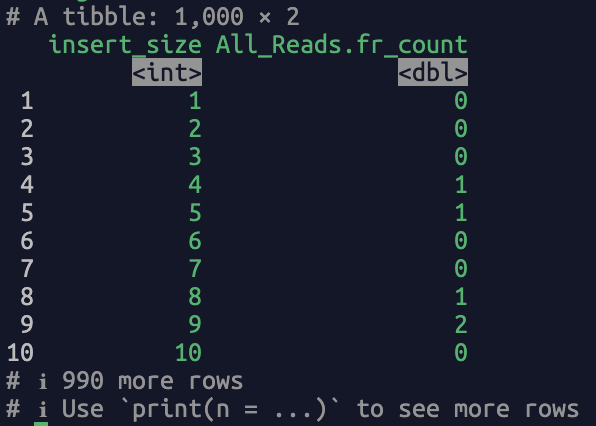
frag_lengths <- read_bam_insert_metrics(bamfile = "/path/to/bamfile.bam")
The returned dataframe contains two columns, i.e., "insert_size" (fragment length) and "All_Reads.fr_count" (the count of the fragment length). A screenshot of the output:
Quick Start 2
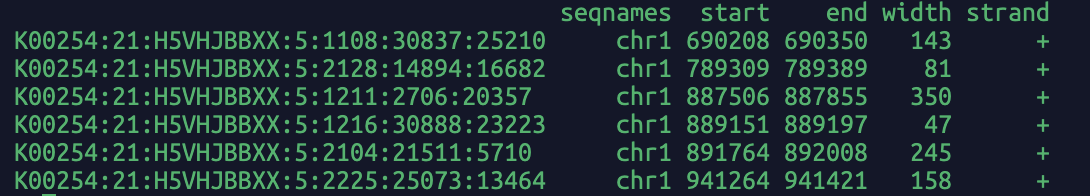
Read bam file, return the fragment name (i.e. read name in bam file) and alignment coordinates in GRanges object in R.
If needed, you can convert the GRanges into a dataframe and the fragment length is stored in the "width" column.
library(cfDNAPro)
# read bam file, do alignment curation
frags <- readBam(bamfile = "/path/to/bamfile.bam")
# convert GRanges object to a dataframe in R
frag_df <- as.data.frame(frags)
A screenshot of the output:
News
cfDNAAPro 1.7.1 (Aug 2024)
- multiple updates
cfDNAPro 1.7.1 (May 2023)
- Resolved issues when building vignette
- Various updates
- Added/Updated readBam() functions
cfDNAPro 1.5.4 (Nov 2022)
- In addition to "bam" and "picard" files as the input, now we accept
"cfdnapro" as input_type to various functions, this 'cfdnapro' input is exactly
the output of
read_bam_insert_metrics function in cfDNAPro package. It is a
tsv file containing two columns, i.e., "insert_size" (fragment length) and
"All_Reads.fr_count" (the count of the fragment length).
cfDNAPro 1.5.3 (Oct 2022)
- added support for hg38-NCBI version, i.e. GRCh38
cfDNAPro 0.99.3 (July 2021)
- Modified vignette.
cfDNAPro 0.99.2 (July 2021)
- Modified vignette.
cfDNAPro 0.99.1 (May 2021)
- Added 'cfDNAPro' into the "watched tag".
cfDNAPro 0.99.0 (May 2021)
- Now cfDNAPro supports bam file as input for data characterisation.
- Coding style improvements.
- Documentation improvements.
- Submitted to Bioconductor.
Installation
Please install our latest version(highly recommended):
if (!require(devtools)) install.packages("devtools")
library(devtools)
devtools::install_github("hw538/cfDNAPro", build_vignettes = TRUE, dependencies = TRUE)
# run below instead if you don't want to build vignettes inside R
# devtools::install_github("hw538/cfDNAPro", build_vignettes = FALSE, dependencies = FALSE)
Or install the released/steady version (i.e., not newest version, some functions might be missing in comparison to functions shown in this webpage)
via Bioconductor:
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("cfDNAPro")
Vignettes/tutorials
visit: https://cfdnapro.readthedocs.io/en/latest/
Citation
Please cite package ‘cfDNAPro’ in publications:
Haichao Wang, Paulius Mennea, Elkie Chan, Hui Zhao, Christopher G. Smith, Tomer Kaplan, Florian Markowetz, Nitzan Rosenfeld(2024). cfDNAPro:An R/Bioconductor package to extract and visualise cell-free DNA biological features. R package version 1.7.1 https://github.com/hw538/cfDNAPro
hw538/cfDNAPro documentation built on Aug. 13, 2024, 2:58 p.m.
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
cfDNAPro
![]()
Official tutorials
For detailed documentation please visit: https://cfdnapro.readthedocs.io/en/latest/
Declaration
cfDNAPro is designed for research only.
Challenges in the cfDNA fragment length calculation
Unlike genomic DNA, cfDNA has specific fragmentation patterns. Ambiguous definition of "fragment length" by various alignment software is raising concerns: see page 9 footnote in SAM file format spec: https://samtools.github.io/hts-specs/SAMv1.pdf Cell-free DNA data fragmentomic analysis requires single-molecule level resolution, which further emphasizes the importance of accurate/un-biased feature extraction.
cfDNAPro is designed to resolve this issue and standardize the cfDNA fragmentomic analysis using the bioconductor R ecosystem.
Input
cfDNAPro is specifically written for cell-free DNA paire-ed whole-genome sequencing data.
Its ensures accurate (i.e. up-to-standard) calculation of fragmentomic features (e.g., fragment lengths and motif)
Output
cfDNApro can extract (i.e., "quantify in a standandised and robust way") these features/bio-markers:
- fragment length
- fragment start/end/upstream/downstream motifs
- copy number variation
- single nucleotide mutation
- more...
Feature extration depends on essential data objects/R packages in the Bioconductor ecosystem, such as Rsamtools, plyranges, GenomicAlignments, GenomeInfoDb and Biostrings.
Data engineering depends on packges in the tidyverse ecosystem, such as dplyr, and stringr.
All plots depend on ggplot2 R packge.
For issues/feature request etc., please contact: Author: Haichao Wang wanghaichao2014@gmail.com Author: Paulius D. Mennea paulius.mennea@cruk.cam.ac.uk Nitzan Rosenfeld Lab admin mailbox: Rosenfeld.LabAdmin@cruk.cam.ac.uk
Quick Start 1
Read in bam file, return the fragment length counts. A straightforward and frequent user case: calculate the fragment size of a bam file, use the following code:
# install cfDNAPro newest version
if (!require(devtools)) install.packages("devtools")
devtools::install_github("hw538/cfDNAPro", build_vignettes = FALSE)
# calculate insert size of a bam file
library(cfDNAPro)
frag_lengths <- read_bam_insert_metrics(bamfile = "/path/to/bamfile.bam")
The returned dataframe contains two columns, i.e., "insert_size" (fragment length) and "All_Reads.fr_count" (the count of the fragment length). A screenshot of the output:
Quick Start 2
Read bam file, return the fragment name (i.e. read name in bam file) and alignment coordinates in GRanges object in R. If needed, you can convert the GRanges into a dataframe and the fragment length is stored in the "width" column.
library(cfDNAPro)
# read bam file, do alignment curation
frags <- readBam(bamfile = "/path/to/bamfile.bam")
# convert GRanges object to a dataframe in R
frag_df <- as.data.frame(frags)
A screenshot of the output:
News
cfDNAAPro 1.7.1 (Aug 2024)
- multiple updates
cfDNAPro 1.7.1 (May 2023)
- Resolved issues when building vignette
- Various updates
- Added/Updated readBam() functions
cfDNAPro 1.5.4 (Nov 2022)
- In addition to "bam" and "picard" files as the input, now we accept
"cfdnapro" as input_type to various functions, this 'cfdnapro' input is exactly
the output of
read_bam_insert_metricsfunction in cfDNAPro package. It is a tsv file containing two columns, i.e., "insert_size" (fragment length) and "All_Reads.fr_count" (the count of the fragment length).
cfDNAPro 1.5.3 (Oct 2022)
- added support for hg38-NCBI version, i.e. GRCh38
cfDNAPro 0.99.3 (July 2021)
- Modified vignette.
cfDNAPro 0.99.2 (July 2021)
- Modified vignette.
cfDNAPro 0.99.1 (May 2021)
- Added 'cfDNAPro' into the "watched tag".
cfDNAPro 0.99.0 (May 2021)
- Now cfDNAPro supports bam file as input for data characterisation.
- Coding style improvements.
- Documentation improvements.
- Submitted to Bioconductor.
Installation
Please install our latest version(highly recommended):
if (!require(devtools)) install.packages("devtools")
library(devtools)
devtools::install_github("hw538/cfDNAPro", build_vignettes = TRUE, dependencies = TRUE)
# run below instead if you don't want to build vignettes inside R
# devtools::install_github("hw538/cfDNAPro", build_vignettes = FALSE, dependencies = FALSE)
Or install the released/steady version (i.e., not newest version, some functions might be missing in comparison to functions shown in this webpage) via Bioconductor:
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("cfDNAPro")
Vignettes/tutorials
visit: https://cfdnapro.readthedocs.io/en/latest/
Citation
Please cite package ‘cfDNAPro’ in publications:
Haichao Wang, Paulius Mennea, Elkie Chan, Hui Zhao, Christopher G. Smith, Tomer Kaplan, Florian Markowetz, Nitzan Rosenfeld(2024). cfDNAPro:An R/Bioconductor package to extract and visualise cell-free DNA biological features. R package version 1.7.1 https://github.com/hw538/cfDNAPro
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
Embedding an R snippet on your website
Add the following code to your website.
For more information on customizing the embed code, read Embedding Snippets.