README.md
In SydneyBioX/scFeatures: scFeatures: Multi-view representations of single-cell and spatial data for disease outcome prediction
scFeatures: Multi-view representations of single-cell and spatial data for disease outcome prediction

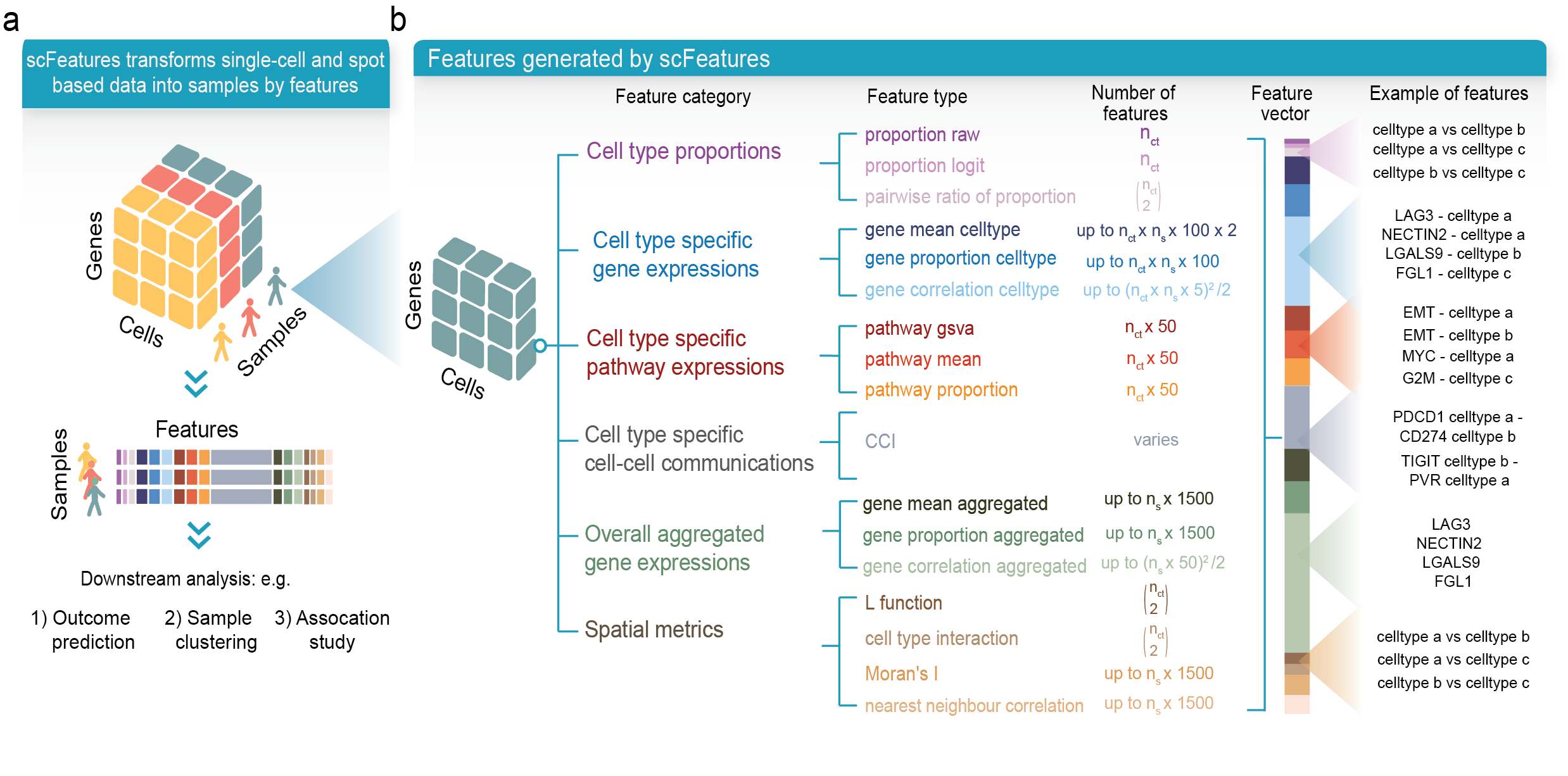
scFeatures is a tool that generates multi-view representations of single-cell and spatial data through the construction of a total of 17 feature types belonging to the following six categories.
- cell type proportions
- cell type specific gene expressions
- cell type specific pathway expressions
- cell type specific cell-cell interaction (CCI) scores
- overall aggregated gene expressions
- spatial metrics
Installation
The latest scFeatures can be installed using devtools:
library(devtools)
devtools::install_github("SydneyBioX/scFeatures")
Quick start
scFeatures can be run using one line of code
scfeatures_result <- scFeatures(data = data, sample = sample, celltype = celltype)
which generates a list of dataframes containing all feature types in the form of samples x features.
Currently, scFeatures support scRNA-seq, spatial proteomics and spatial transcriptomics.
For scRNA-seq, run:
data("example_scrnaseq" , package = "scFeatures")
data <- example_scrnaseq
scfeatures_result <- scFeatures(data = data@assays$RNA@data,
sample = data$sample,
celltype = data$celltype,
type = "scrna",
ncores = 8,
species = "Homo sapiens")
For spatial proteomics, run:
# note, spatial data requires spatial coordinates of each cell.
spatialCoords <- list( sample( 1:ncol(data), ncol(data)) ,
sample( 1:ncol(data), ncol(data) )) # generate fake coordinates
scfeatures_result <- scFeatures(data = data@assays$RNA@data,
sample = data$sample,
celltype = data$celltype,
type = "spatial_p",
spatialCoords = spatialCoords,
ncores = 8,
species = "Homo sapiens")
For spatial transcriptomics, run:
# note, spatial data requires spatial coordinates of each cell.
spatialCoords <- list( sample( 1:ncol(data), ncol(data)) ,
sample( 1:ncol(data), ncol(data) ))
# as well as predicted probability of cell types in each spot
spotProbability <- t(gtools::rdirichlet( ncol(data), rep(1, 5))) # simulate the cell type prediction result based on 5 cell types
rownames( spotProbability) <- c("Cell type A", "Cell type B" , "Cell type C",
"Cell type D", "Cell type E")
colnames( spotProbability ) <- colnames(data)
scfeatures_result <- scFeatures(data = data@assays$RNA@data,
sample = data$sample,
celltype = data$celltype,
type = "spatial_t",
spatialCoords = spatialCoords,
spotProbability = spotProbability,
ncores = 8,
species = "Homo sapiens")
Detailed vignette
Please see https://sydneybiox.github.io/scFeatures/articles/scFeatures_overview.html.
Reference
Cao, Y., Lin, Y., Patrick, E., Yang, P., & Yang, J. Y. H. (2022). scFeatures: multi-view representations of single-cell and spatial data for disease outcome prediction. In O. Vitek (Ed.), Bioinformatics (Vol. 38, Issue 20, pp. 4745–4753). Oxford University Press (OUP). https://doi.org/10.1093/bioinformatics/btac590
SydneyBioX/scFeatures documentation built on Aug. 29, 2024, 10:37 a.m.
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
scFeatures: Multi-view representations of single-cell and spatial data for disease outcome prediction
scFeatures is a tool that generates multi-view representations of single-cell and spatial data through the construction of a total of 17 feature types belonging to the following six categories.
- cell type proportions
- cell type specific gene expressions
- cell type specific pathway expressions
- cell type specific cell-cell interaction (CCI) scores
- overall aggregated gene expressions
- spatial metrics
Installation
The latest scFeatures can be installed using devtools:
library(devtools)
devtools::install_github("SydneyBioX/scFeatures")
Quick start
scFeatures can be run using one line of code
scfeatures_result <- scFeatures(data = data, sample = sample, celltype = celltype)
which generates a list of dataframes containing all feature types in the form of samples x features.
Currently, scFeatures support scRNA-seq, spatial proteomics and spatial transcriptomics.
For scRNA-seq, run:
data("example_scrnaseq" , package = "scFeatures")
data <- example_scrnaseq
scfeatures_result <- scFeatures(data = data@assays$RNA@data,
sample = data$sample,
celltype = data$celltype,
type = "scrna",
ncores = 8,
species = "Homo sapiens")
For spatial proteomics, run:
# note, spatial data requires spatial coordinates of each cell.
spatialCoords <- list( sample( 1:ncol(data), ncol(data)) ,
sample( 1:ncol(data), ncol(data) )) # generate fake coordinates
scfeatures_result <- scFeatures(data = data@assays$RNA@data,
sample = data$sample,
celltype = data$celltype,
type = "spatial_p",
spatialCoords = spatialCoords,
ncores = 8,
species = "Homo sapiens")
For spatial transcriptomics, run:
# note, spatial data requires spatial coordinates of each cell.
spatialCoords <- list( sample( 1:ncol(data), ncol(data)) ,
sample( 1:ncol(data), ncol(data) ))
# as well as predicted probability of cell types in each spot
spotProbability <- t(gtools::rdirichlet( ncol(data), rep(1, 5))) # simulate the cell type prediction result based on 5 cell types
rownames( spotProbability) <- c("Cell type A", "Cell type B" , "Cell type C",
"Cell type D", "Cell type E")
colnames( spotProbability ) <- colnames(data)
scfeatures_result <- scFeatures(data = data@assays$RNA@data,
sample = data$sample,
celltype = data$celltype,
type = "spatial_t",
spatialCoords = spatialCoords,
spotProbability = spotProbability,
ncores = 8,
species = "Homo sapiens")
Detailed vignette
Please see https://sydneybiox.github.io/scFeatures/articles/scFeatures_overview.html.
Reference
Cao, Y., Lin, Y., Patrick, E., Yang, P., & Yang, J. Y. H. (2022). scFeatures: multi-view representations of single-cell and spatial data for disease outcome prediction. In O. Vitek (Ed.), Bioinformatics (Vol. 38, Issue 20, pp. 4745–4753). Oxford University Press (OUP). https://doi.org/10.1093/bioinformatics/btac590
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
Embedding an R snippet on your website
Add the following code to your website.
For more information on customizing the embed code, read Embedding Snippets.