Nothing
README.md
In scRecover: scRecover for imputation of single-cell RNA-seq data
scRecover
Zhun Miao, Xuegong Zhang
2019-04-28

1. Introduction
scRecover is an R package for imputation of single-cell RNA-seq (scRNA-seq) data. It will detect and impute dropout values in a scRNA-seq raw read counts matrix while keeping the real zeros unchanged.
Since there are both dropout zeros and real zeros in scRNA-seq data, imputation methods should not impute all the zeros to non-zero values. To distinguish dropout and real zeros, scRecover employs the Zero-Inflated Negative Binomial (ZINB) model for dropout probability estimation of each gene and accumulation curves for prediction of dropout number in each cell. By combination with scImpute, SAVER and MAGIC, it not only detects dropout and real zeros at higher accuracy, but also improve the downstream clustering and visualization results.
2. Citation
If you use scRecover in published research, please cite:
3. Installation
To install scRecover from Bioconductor:
```{r Installation from Bioconductor, eval = FALSE}
if(!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("scRecover")
To install the *developmental version* from [**Bioconductor**](https://bioconductor.org/packages/devel/bioc/html/scRecover.html):
```{r Developmental version from Bioconductor, eval = FALSE}
if(!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("scRecover", version = "devel")
Or install the developmental version from GitHub:
```{r Installation from GitHub, eval = FALSE}
if(!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("miaozhun/scRecover")
To load **`scRecover`** and other required packages for the vignettes in R:
```{r Load scRecover, eval = TRUE}
library(scRecover)
library(BiocParallel)
suppressMessages(library(SingleCellExperiment))
4. Input
scRecover takes two inputs: counts and one of Kcluster or labels.
The input counts is a scRNA-seq read counts matrix or a SingleCellExperiment object which contains the read counts matrix. The rows of the matrix are genes and columns are cells.
Kcluster is an integer specifying the number of cell subpopulations. This parameter can be determined based on prior knowledge or clustering of raw data. Kcluster is used to determine the candidate neighbors of each cell and need not to be very accurate.
labels is a character/integer vector specifying the cell type of each column in the raw count matrix. Only needed when Kcluster = NULL. Each cell type should have at least two cells for imputation.
5. Test data
Users can load the test data in scRecover by
```{r Load scRecoverTest}
data(scRecoverTest)
The test data `counts` in `scRecoverTest` is a scRNA-seq read counts matrix which has 200 genes (rows) and 150 cells (columns).
```{r counts}
dim(counts)
counts[1:6, 1:6]
The object labels in scRecoverTest is a vector of integer specifying the cell types in the read counts matrix, corresponding to the columns of counts.
```{r labels}
length(labels)
table(labels)
The object `oneCell` in `scRecoverTest` is a vector of a cell's raw read counts for each gene.
```{r oneCell}
head(oneCell)
length(oneCell)
6. Usage
6.1 Imputation using scRecover
6.1.1 With read counts matrix input
Here is an example to run scRecover with read counts matrix input:
```{r demo1, eval = TRUE}
Load test data for scRecover
data(scRecoverTest)
Run scRecover with Kcluster specified
scRecover(counts = counts, Kcluster = 2, outputDir = "./outDir_scRecover/", verbose = FALSE)
Or run scRecover with labels specified
scRecover(counts = counts, labels = labels, outputDir = "./outDir_scRecover/")
### 6.1.2 With SingleCellExperiment input
The [`SingleCellExperiment`](http://bioconductor.org/packages/SingleCellExperiment/) class is a widely used S4 class for storing single-cell genomics data. **`scRecover`** also could take the `SingleCellExperiment` data representation as input.
Here is an example to run **`scRecover`** with `SingleCellExperiment` input:
```{r demo2, eval = TRUE}
# Load test data for scRecover
data(scRecoverTest)
# Convert the test data in scRecover to SingleCellExperiment data representation
sce <- SingleCellExperiment(assays = list(counts = as.matrix(counts)))
# Run scRecover with SingleCellExperiment input sce (Kcluster specified)
scRecover(counts = sce, Kcluster = 2, outputDir = "./outDir_scRecover/", verbose = FALSE)
# Or run scRecover with SingleCellExperiment input sce (labels specified)
# scRecover(counts = sce, labels = labels, outputDir = "./outDir_scRecover/")
6.2 Estimate dropout gene number in a cell
Function estDropoutNum in the package could estimate the dropout gene number or all expressed gene number (namely observed gene number plus dropout gene number) in a cell:
```{r demo3, eval = TRUE}
Load test data
data(scRecoverTest)
Downsample 10% read counts in oneCell
set.seed(999)
oneCell.down <- countsSampling(counts = oneCell, fraction = 0.1)
Count the groundtruth dropout gene number in the downsampled cell
sum(oneCell.down == 0 & oneCell != 0)
Estimate the dropout gene number in the downsampled cell by estDropoutNum
estDropoutNum(sample = oneCell.down, depth = 10, return = "dropoutNum")
Blow shows the expressed gene number predicted by `estDropoutNum` with 10% downsampled reads and the groundtruth expressed gene number derived by downsampling when the reads depth varying from 0% to 100% of the total reads.

# 7. Output
Imputed expression matrices of **`scRecover`** will be saved in the output directory specified by \code{outputDir} or a folder named with prefix 'outDir_scRecover_' under the current working directory when \code{outputDir} is unspecified.
# 8. Parallelization
**`scRecover`** integrates parallel computing function with [`BiocParallel`](http://bioconductor.org/packages/BiocParallel/) package. Users could just set `parallel = TRUE` (default) in function `scRecover` to enable parallelization and leave the `BPPARAM` parameter alone.
```{r demo4, eval = FALSE}
# Run scRecover with Kcluster specified
scRecover(counts = counts, Kcluster = 2, parallel = TRUE)
# Run scRecover with labels specified
scRecover(counts = counts, labels = labels, parallel = TRUE)
Advanced users could use a BiocParallelParam object from package BiocParallel to fill in the BPPARAM parameter to specify the parallel back-end to be used and its configuration parameters.
8.1 For Unix and Mac users
The best choice for Unix and Mac users is to use MulticoreParam to configure a multicore parallel back-end:
```{r demo5, eval = FALSE}
Set the parameters and register the back-end to be used
param <- MulticoreParam(workers = 18, progressbar = TRUE)
register(param)
Run scRecover with 18 cores (Kcluster specified)
scRecover(counts = counts, Kcluster = 2, parallel = TRUE, BPPARAM = param)
Run scRecover with 18 cores (labels specified)
scRecover(counts = counts, labels = labels, parallel = TRUE, BPPARAM = param)
## 8.2 For Windows users
For Windows users, use `SnowParam` to configure a Snow back-end is a good choice:
```{r demo6, eval = FALSE}
# Set the parameters and register the back-end to be used
param <- SnowParam(workers = 8, type = "SOCK", progressbar = TRUE)
register(param)
# Run scRecover with 8 cores (Kcluster specified)
scRecover(counts = counts, Kcluster = 2, parallel = TRUE, BPPARAM = param)
# Run scRecover with 8 cores (labels specified)
scRecover(counts = counts, labels = labels, parallel = TRUE, BPPARAM = param)
See the Reference Manual of BiocParallel package for more details of the BiocParallelParam class.
9. Evaluation of scRecover
9.1 On downsampling data
We evaluated SAVER, scImpute, MAGIC and their combined with scRecover version SAVER+scRecover, scImpute+scRecover, MAGIC+scRecover on a downsampling scRNA-seq dataset generated by random sampling of reads from a SMART-seq2 scRNA-seq dataset (Petropoulos S, et al. Cell, 2016, https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-3929/).
9.1.1 Accuracy of dropout prediction
We found after combined with scRecover, scImpute+scRecover, SAVER+scRecover and MAGIC+scRecover will have higher accuracy than scImpute, SAVER and MAGIC respectively.

9.1.2 Predicted dropout number
We found scImpute+scRecover, SAVER+scRecover and MAGIC+scRecover will have predicted dropout numbers closer to the real dropout number than without combination with scRecover.

9.2 On 10X data
We applied the 6 imputation methods to a 10X scRNA-seq dataset (https://support.10xgenomics.com/single-cell-gene-expression/datasets/3.0.0/heart_1k_v3).
Then we measured the downstream clustering and visualization results by comparing to the cell labels originated from the dataset and deriving their Adjusted Rand Index (ARI) and Jaccard indexes (the larger, the better).
We found a significant improvement of SAVER, scImpute and MAGIC after combined with scRecover.

Gene number before and after imputation:
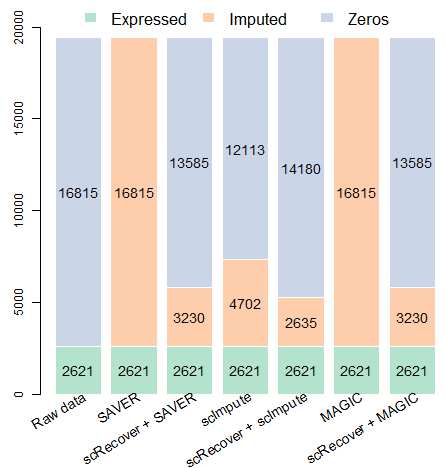
9.3 On SMART-seq data
Next, we applied the 6 imputation methods to a SMART-seq scRNA-seq dataset (Chu L, et al. Genome Biology, 2016, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE75748).
Then we measured the downstream clustering and visualization results by comparing to the cell labels originated from the dataset and deriving their Adjusted Rand Index (ARI) and Jaccard indexes (the larger, the better).
We found a significant improvement of SAVER, scImpute and MAGIC after combined with scRecover.

Gene number before and after imputation:

10. Help
Use browseVignettes("scRecover") to see the vignettes of scRecover in R after installation.
Use the following code in R to get access to the help documentation for scRecover:
```{r help, eval = FALSE}
Documentation for scRecover
?scRecover
Documentation for estDropoutNum
?estDropoutNum
Documentation for countsSampling
?countsSampling
Documentation for normalization
?normalization
Documentation for test data
?scRecoverTest
?counts
?labels
?oneCell
```
You are also welcome to contact the author by email for help.
11. Author
Zhun Miao, Xuegong Zhang <zhangxg@tsinghua.edu.cn>
MOE Key Laboratory of Bioinformatics; Bioinformatics Division and Center for Synthetic & Systems Biology, TNLIST; Department of Automation, Tsinghua University, Beijing 100084, China.
Try the scRecover package in your browser
Any scripts or data that you put into this service are public.
scRecover documentation built on Nov. 8, 2020, 5:03 p.m.
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
scRecover
Zhun Miao, Xuegong Zhang
2019-04-28
![]()
1. Introduction
scRecover is an R package for imputation of single-cell RNA-seq (scRNA-seq) data. It will detect and impute dropout values in a scRNA-seq raw read counts matrix while keeping the real zeros unchanged.
Since there are both dropout zeros and real zeros in scRNA-seq data, imputation methods should not impute all the zeros to non-zero values. To distinguish dropout and real zeros, scRecover employs the Zero-Inflated Negative Binomial (ZINB) model for dropout probability estimation of each gene and accumulation curves for prediction of dropout number in each cell. By combination with scImpute, SAVER and MAGIC, it not only detects dropout and real zeros at higher accuracy, but also improve the downstream clustering and visualization results.
2. Citation
If you use scRecover in published research, please cite:
3. Installation
To install scRecover from Bioconductor:
```{r Installation from Bioconductor, eval = FALSE} if(!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager") BiocManager::install("scRecover")
To install the *developmental version* from [**Bioconductor**](https://bioconductor.org/packages/devel/bioc/html/scRecover.html):
```{r Developmental version from Bioconductor, eval = FALSE}
if(!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("scRecover", version = "devel")
Or install the developmental version from GitHub:
```{r Installation from GitHub, eval = FALSE} if(!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager") BiocManager::install("miaozhun/scRecover")
To load **`scRecover`** and other required packages for the vignettes in R:
```{r Load scRecover, eval = TRUE}
library(scRecover)
library(BiocParallel)
suppressMessages(library(SingleCellExperiment))
4. Input
scRecover takes two inputs: counts and one of Kcluster or labels.
The input counts is a scRNA-seq read counts matrix or a SingleCellExperiment object which contains the read counts matrix. The rows of the matrix are genes and columns are cells.
Kcluster is an integer specifying the number of cell subpopulations. This parameter can be determined based on prior knowledge or clustering of raw data. Kcluster is used to determine the candidate neighbors of each cell and need not to be very accurate.
labels is a character/integer vector specifying the cell type of each column in the raw count matrix. Only needed when Kcluster = NULL. Each cell type should have at least two cells for imputation.
5. Test data
Users can load the test data in scRecover by
```{r Load scRecoverTest} data(scRecoverTest)
The test data `counts` in `scRecoverTest` is a scRNA-seq read counts matrix which has 200 genes (rows) and 150 cells (columns).
```{r counts}
dim(counts)
counts[1:6, 1:6]
The object labels in scRecoverTest is a vector of integer specifying the cell types in the read counts matrix, corresponding to the columns of counts.
```{r labels} length(labels) table(labels)
The object `oneCell` in `scRecoverTest` is a vector of a cell's raw read counts for each gene.
```{r oneCell}
head(oneCell)
length(oneCell)
6. Usage
6.1 Imputation using scRecover
6.1.1 With read counts matrix input
Here is an example to run scRecover with read counts matrix input:
```{r demo1, eval = TRUE}
Load test data for scRecover
data(scRecoverTest)
Run scRecover with Kcluster specified
scRecover(counts = counts, Kcluster = 2, outputDir = "./outDir_scRecover/", verbose = FALSE)
Or run scRecover with labels specified
scRecover(counts = counts, labels = labels, outputDir = "./outDir_scRecover/")
### 6.1.2 With SingleCellExperiment input
The [`SingleCellExperiment`](http://bioconductor.org/packages/SingleCellExperiment/) class is a widely used S4 class for storing single-cell genomics data. **`scRecover`** also could take the `SingleCellExperiment` data representation as input.
Here is an example to run **`scRecover`** with `SingleCellExperiment` input:
```{r demo2, eval = TRUE}
# Load test data for scRecover
data(scRecoverTest)
# Convert the test data in scRecover to SingleCellExperiment data representation
sce <- SingleCellExperiment(assays = list(counts = as.matrix(counts)))
# Run scRecover with SingleCellExperiment input sce (Kcluster specified)
scRecover(counts = sce, Kcluster = 2, outputDir = "./outDir_scRecover/", verbose = FALSE)
# Or run scRecover with SingleCellExperiment input sce (labels specified)
# scRecover(counts = sce, labels = labels, outputDir = "./outDir_scRecover/")
6.2 Estimate dropout gene number in a cell
Function estDropoutNum in the package could estimate the dropout gene number or all expressed gene number (namely observed gene number plus dropout gene number) in a cell:
```{r demo3, eval = TRUE}
Load test data
data(scRecoverTest)
Downsample 10% read counts in oneCell
set.seed(999) oneCell.down <- countsSampling(counts = oneCell, fraction = 0.1)
Count the groundtruth dropout gene number in the downsampled cell
sum(oneCell.down == 0 & oneCell != 0)
Estimate the dropout gene number in the downsampled cell by estDropoutNum
estDropoutNum(sample = oneCell.down, depth = 10, return = "dropoutNum")
Blow shows the expressed gene number predicted by `estDropoutNum` with 10% downsampled reads and the groundtruth expressed gene number derived by downsampling when the reads depth varying from 0% to 100% of the total reads.

# 7. Output
Imputed expression matrices of **`scRecover`** will be saved in the output directory specified by \code{outputDir} or a folder named with prefix 'outDir_scRecover_' under the current working directory when \code{outputDir} is unspecified.
# 8. Parallelization
**`scRecover`** integrates parallel computing function with [`BiocParallel`](http://bioconductor.org/packages/BiocParallel/) package. Users could just set `parallel = TRUE` (default) in function `scRecover` to enable parallelization and leave the `BPPARAM` parameter alone.
```{r demo4, eval = FALSE}
# Run scRecover with Kcluster specified
scRecover(counts = counts, Kcluster = 2, parallel = TRUE)
# Run scRecover with labels specified
scRecover(counts = counts, labels = labels, parallel = TRUE)
Advanced users could use a BiocParallelParam object from package BiocParallel to fill in the BPPARAM parameter to specify the parallel back-end to be used and its configuration parameters.
8.1 For Unix and Mac users
The best choice for Unix and Mac users is to use MulticoreParam to configure a multicore parallel back-end:
```{r demo5, eval = FALSE}
Set the parameters and register the back-end to be used
param <- MulticoreParam(workers = 18, progressbar = TRUE) register(param)
Run scRecover with 18 cores (Kcluster specified)
scRecover(counts = counts, Kcluster = 2, parallel = TRUE, BPPARAM = param)
Run scRecover with 18 cores (labels specified)
scRecover(counts = counts, labels = labels, parallel = TRUE, BPPARAM = param)
## 8.2 For Windows users
For Windows users, use `SnowParam` to configure a Snow back-end is a good choice:
```{r demo6, eval = FALSE}
# Set the parameters and register the back-end to be used
param <- SnowParam(workers = 8, type = "SOCK", progressbar = TRUE)
register(param)
# Run scRecover with 8 cores (Kcluster specified)
scRecover(counts = counts, Kcluster = 2, parallel = TRUE, BPPARAM = param)
# Run scRecover with 8 cores (labels specified)
scRecover(counts = counts, labels = labels, parallel = TRUE, BPPARAM = param)
See the Reference Manual of BiocParallel package for more details of the BiocParallelParam class.
9. Evaluation of scRecover
9.1 On downsampling data
We evaluated SAVER, scImpute, MAGIC and their combined with scRecover version SAVER+scRecover, scImpute+scRecover, MAGIC+scRecover on a downsampling scRNA-seq dataset generated by random sampling of reads from a SMART-seq2 scRNA-seq dataset (Petropoulos S, et al. Cell, 2016, https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-3929/).
9.1.1 Accuracy of dropout prediction
We found after combined with scRecover, scImpute+scRecover, SAVER+scRecover and MAGIC+scRecover will have higher accuracy than scImpute, SAVER and MAGIC respectively.
9.1.2 Predicted dropout number
We found scImpute+scRecover, SAVER+scRecover and MAGIC+scRecover will have predicted dropout numbers closer to the real dropout number than without combination with scRecover.
9.2 On 10X data
We applied the 6 imputation methods to a 10X scRNA-seq dataset (https://support.10xgenomics.com/single-cell-gene-expression/datasets/3.0.0/heart_1k_v3).
Then we measured the downstream clustering and visualization results by comparing to the cell labels originated from the dataset and deriving their Adjusted Rand Index (ARI) and Jaccard indexes (the larger, the better).
We found a significant improvement of SAVER, scImpute and MAGIC after combined with scRecover.
Gene number before and after imputation:
9.3 On SMART-seq data
Next, we applied the 6 imputation methods to a SMART-seq scRNA-seq dataset (Chu L, et al. Genome Biology, 2016, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE75748).
Then we measured the downstream clustering and visualization results by comparing to the cell labels originated from the dataset and deriving their Adjusted Rand Index (ARI) and Jaccard indexes (the larger, the better).
We found a significant improvement of SAVER, scImpute and MAGIC after combined with scRecover.
Gene number before and after imputation:
10. Help
Use browseVignettes("scRecover") to see the vignettes of scRecover in R after installation.
Use the following code in R to get access to the help documentation for scRecover:
```{r help, eval = FALSE}
Documentation for scRecover
?scRecover
Documentation for estDropoutNum
?estDropoutNum
Documentation for countsSampling
?countsSampling
Documentation for normalization
?normalization
Documentation for test data
?scRecoverTest ?counts ?labels ?oneCell ```
You are also welcome to contact the author by email for help.
11. Author
Zhun Miao, Xuegong Zhang <zhangxg@tsinghua.edu.cn>
MOE Key Laboratory of Bioinformatics; Bioinformatics Division and Center for Synthetic & Systems Biology, TNLIST; Department of Automation, Tsinghua University, Beijing 100084, China.
Try the scRecover package in your browser
Any scripts or data that you put into this service are public.
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
{kind=link}
Embedding an R snippet on your website
Add the following code to your website.
For more information on customizing the embed code, read Embedding Snippets.